Antimitotic Tubulin Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
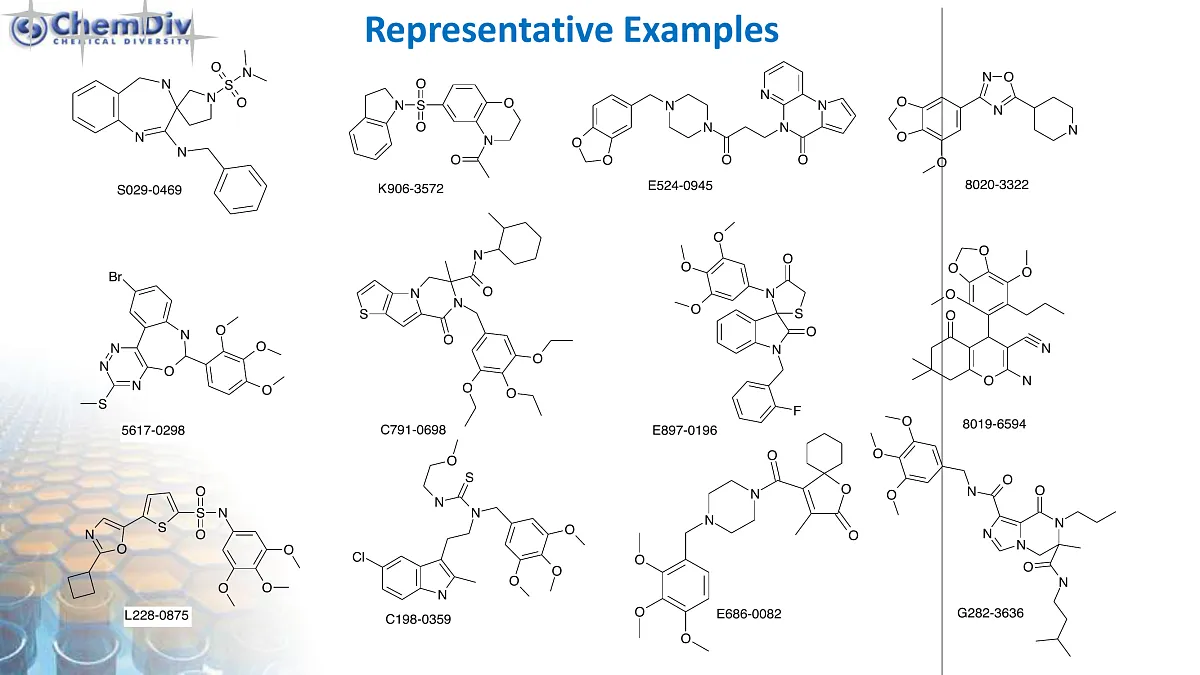
Antimitotic Agents Library
ChemDiv’s Library of Antimitotic Agents comprises 18,000 compounds.
Microtubules are one of the primary components of the cytoskeleton, which is a framework essential for organizing numerous critical cellular processes, including cell shape, cellular polarity, cell motility, signal transduction, cell division, intracellular transport, and muscle contraction. Microtubules present hollow cylindrical protein fibers composed of alternating α- and β-tubulins. They are stacked together through non-covalent bonding, and their cross-section has 13 parallel α- and β-tubulin heterodimers. As integral components of mitotic spindles, microtubules play a critical role in cell proliferation. Dynamic microtubules that make up the mitotic spindles serve as targets for the most antimitotic agents. There are three major classes of these agents: microtubule stabilizing agents, primarily taxanes and epothilones; tubulin polymerization inhibitors that bind at the Vinca alkaloid site; and tubulin polymerization inhibitors that bind at the colchicine site [1].
[1] Q. Li and H. L. Sham, “Discovery and development of antimitotic agents that inhibit tubulin polymerisation for the treatment of cancer,” Expert Opin. Ther. Pat., vol. 12, no. 11, pp. 1663–1702, doi: 10.1517/13543776.12.11.1663.
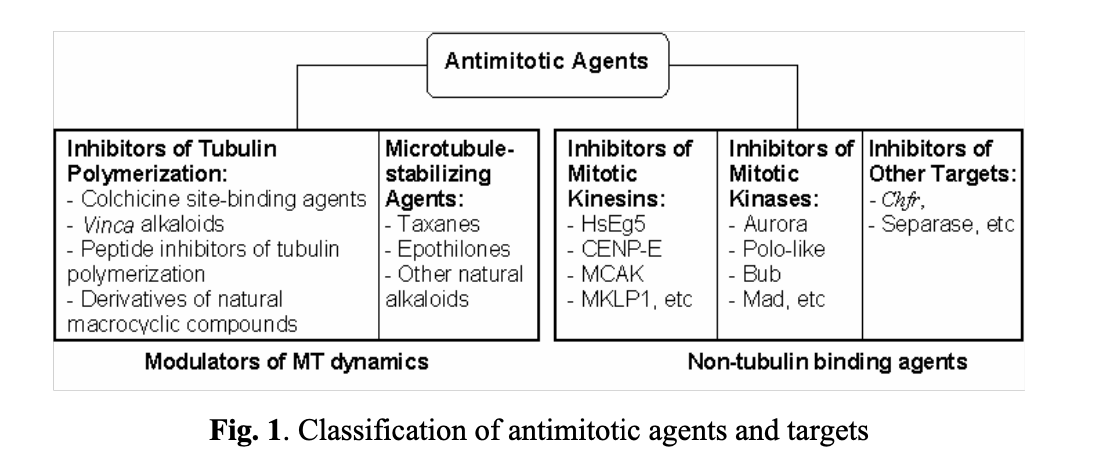
With several successful anticancer drugs already on the market and numerous compounds undergoing clinical development, antimitotic agents constitute a notable category of anticancer therapies. However, the clinical utility of the tubulin-binding drugs is significantly restrained due to multiple drug resistance (MDR), suboptimal pharmacokinetics, and a narrow therapeutic index. Another notable limitation of the current modulators of tubulin dynamics is their marginal clinical efficacy. Although they demonstrate impressive in vitro activity, the majority of tubulin-binding agents have not shown significant antitumor activity in animals or clinical settings. This discrepancy is often attributed to a poor balance between efficacy and toxicity, known as the therapeutic window, which is affected by various drug characteristics, including pharmacokinetics, off-target toxicity, and other less recognized factors. Additionally, drug efflux pumps contribute to tumor resistance against tubulin-binding drugs. Consequently, there is a continuous need for modulators targeting different intracellular components that can achieve the anti-mitotic effects of traditional tubulin binding without notable adverse effects. For instance, kinesins, the microtubule motor proteins, play a critical role in mitotic spindle function and represent potential targets for novel cancer therapy discoveries. Proteins that regulate cellular progression through mitosis, such as kinases and cysteine proteases, including Polo, Bub, Mad, Aurora, Cdk1, separase, and others, also provide avenues for therapeutic intervention.
Historically, researchers have been focusing on two classes of antimitotic agents (see Fig. 1). The first class comprises compounds reversibly binding to tubulin and preventing the assembly and disassembly of microtubules (modulators of MT dynamics). The second class includes molecules that indirectly regulate mitotic events by interacting with specific intracellular targets, such as mitotic kinesins, kinases, separase, and others.
Mitotic kinases
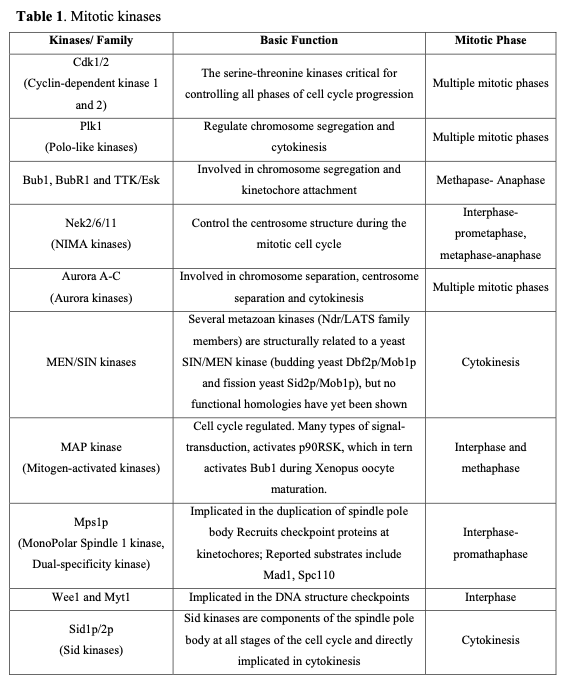
Mitotic failure is one of the critical sources of the genetic instability that characterizes oncogenesis. Clinical evidence indicates that numerous proteins regulating mitosis are aberrantly expressed in human tumors. Mitotic progression regulation relies on two post-translational mechanisms, namely protein phosphorylation and proteolysis. Mitotic kinases mediate protein phosphorylation and serve as major regulators controlling mitosis progression (Table 1).
The mitotic cycle is made up of four major stages (see Fig. 2). To ensure that daughter cells get identical genome copies, the cell cycle progression is stringently regulated and controlled by a cadence of mitotic kinases. The most important one among them are Cdk1, Plk1, and Nek2, which regulate mitotic checkpoints at the interphase and prophase stages. Cdk1 is a key player from prophase through to telophase, and Aurora kinases are critical for the cell progression over all stages of mitosis.
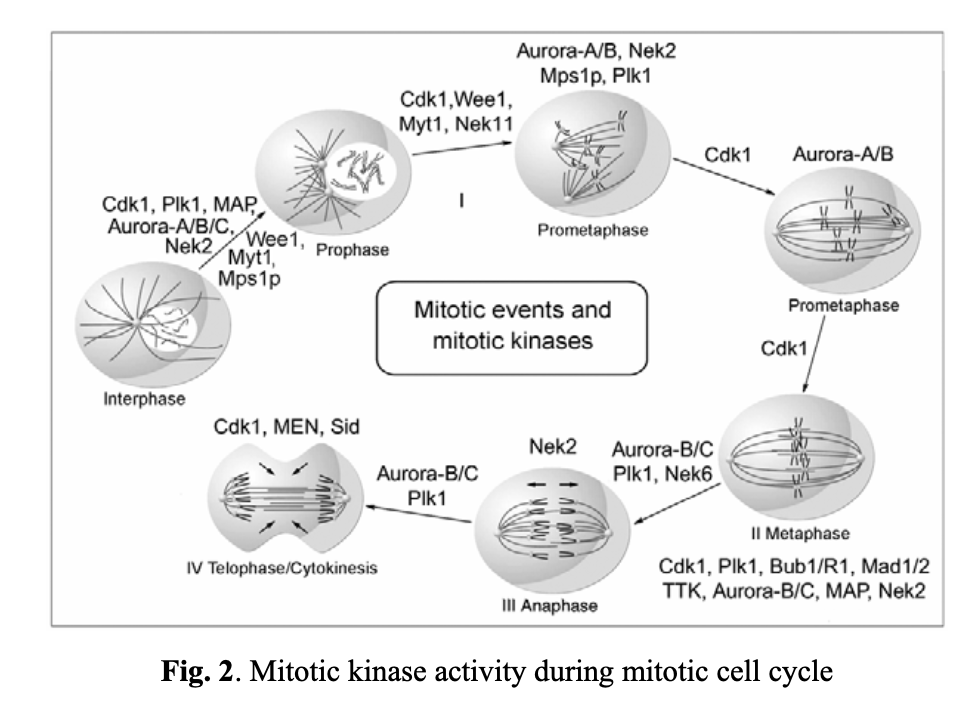
As previously mentioned, mitotic kinases are critical elements in regulating mitotic checkpoints. CDK1, a nonredundant cyclin-dependent kinase (CDK), plays a vital role in cell division, especially at the early stages of mitosis (entry into mitosis). For instance, a markedly reduced CDK1 transcription and/or the loss of its active form (CDK1-P-Thr(161)) is associated with discontinued mitosis in tumor cells. This occurs at the G2/M transition phase when the activity of the dual-specific phosphatase Cdc25C toward Cdk1 exceeds the activity of the opposing kinases Wee1 and Myt1. These proteins, in turn, are regulated by DNA structure checkpoints that delay mitosis onset in the presence of unreplicated or damaged DNA. Cdc25C can be inhibited by two other kinases, Chk1 and Chk2, also involved in the DNA structure checkpoint signaling pathway. Wee1 and Myt1 are upregulated through the same pathways. Notably, the Plk1 kinase also activates Cdc25C.
The spindle assembly checkpoint ensures the accurate segregation of chromosomes during mitosis. It blocks the anaphase stage until all chromosomes are properly attached to a bipolar mitotic spindle. As unstable chromosomes are detected, the spindle checkpoint inhibits the ubiquitin ligase activity of the anaphase-promoting complex or cyclosome (APC/C). This inhibition is mediated by proteins encoded by the BUB and MAD genes. Specifically, mitotic kinases, including Bub1/3, BubR1, Bub3, Mad1, and Mad2, are recruited to unattached kinetochores (see Fig. 3). DNA replication and centrosome duplication are regulated by E2F transcription factors, Mps1p kinases, cyclin A/E, and the Cdc20 protein. Mitotic checkpoint protein complexes, composed of BubR1, Bub3, and Mad2, bind to and inhibit APC/Cdc20 until all chromosomes are properly attached to the mitotic spindle and aligned on the metaphase plate (Fig. 3A, 3B, and 3C).
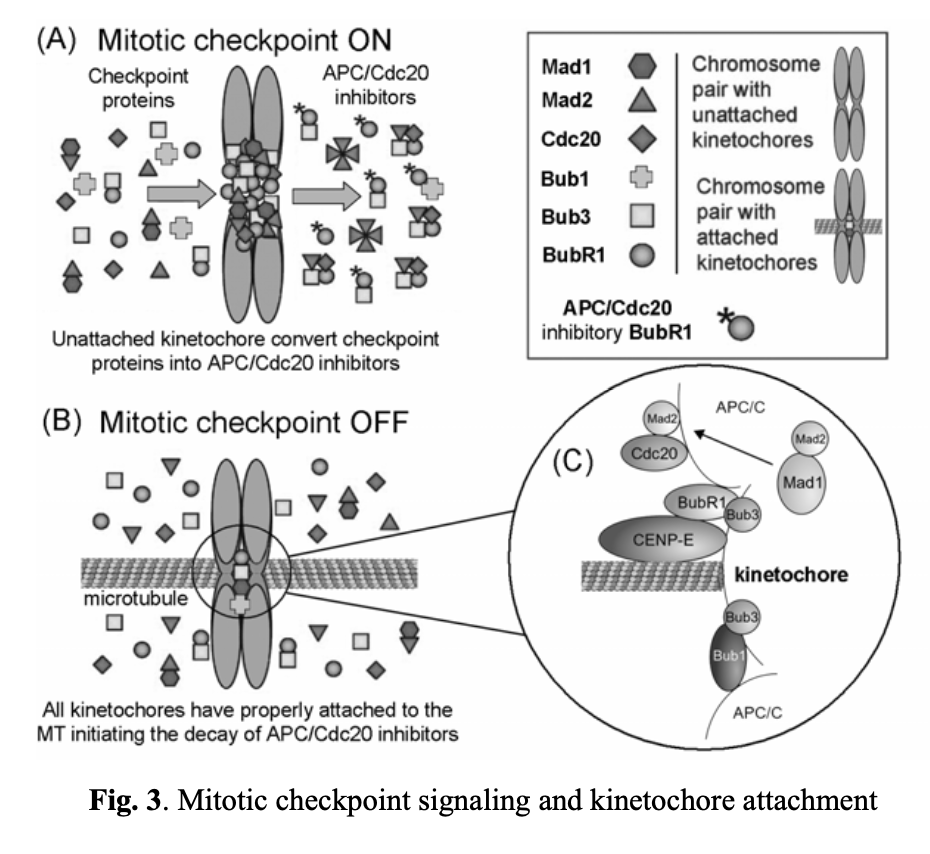
The ability of a cell to track its temporal and spatial fidelity during progression through the cell cycle is essential for survival. A spindle-positioning checkpoint has been initially described in the yeast S. cerevisiaexi. The first identified component of this step was Bub2/Bfa1 GTPase-activating protein (GAP). It is responsible for keeping small GTPase (Tem1p) inactive until the spindle is properly oriented. The net result is inhibition of the mitotic exit network (MEN) activationxii. The signaling cascade that is responsible for initiating MEN includes mitotic kinases that activate Cdc14p phosphatase. Cdc14p activates APC/C/Cdh1 complex, dephosphorylates Cdk1-inhibitor Sic1p (causing its stabilization) and transcription factor Swi5p (enhancing the production of Sic1p)xiii. These events lead to destruction of mitotic cyclin–CDK complexes only when the spindle-positioning checkpoint is satisfied.
Small molecule inhibitors of mitotic kinases
Considering the pivotal role of protein phosphorylation in mitotic checkpoints, spindle function, and chromosome segregation, it is not surprising that several mitotic kinases have been implicated in tumorigenesis. For example, CDK1-8, Aurora (Aur) (A, B, C), CDK (Cdk1, 2), Polo-like (Plk1-4), Nek (NIMA1-11), Bub (Bub1, BubR1) and other kinases are implicated in the mediation of centrosome duplication, chromosome segregation, and cytokinesis in diverse human tumors. These enzymes also regulate the centrosome cycle, spindle checkpoint, microtubule-kinetochore attachment, spindle assembly, and chromosome condensation. Several potent and selective inhibitors of mitotic kinases have entered clinical trials (Table 2).
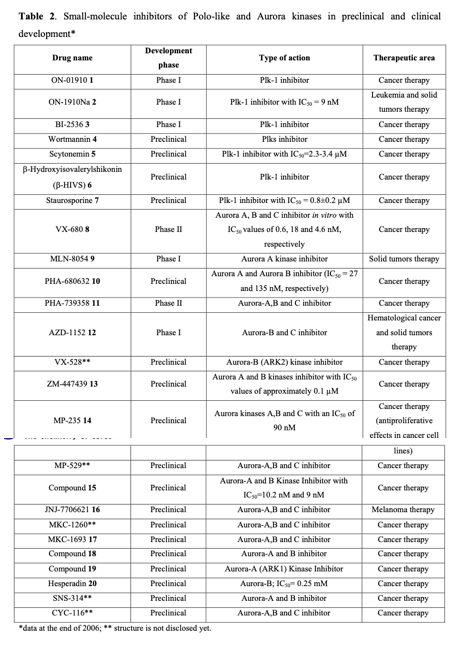
Inhibitors of Polo-like kinases
Polo-like kinases (Plks) belong to a family of conserved serine/threonine kinases with a polo-box domain that have similar but non-redundant functions in cell cycle progression, controlling the mitotic entry of proliferating cells and regulating various mitosis factors required for successful cytokinesis. For example, they are essential for the activities of the MT organization center. They are critical elements in mitotic entry, spindle formation, and cytokinesis. Multiple Plks are found in mammalian cells (Plk-1, Plk2/Snk, Plk3/Fnk/Prk, and Plk4/Sak) as well as in Xenopus (Plx1- 3). Among the four known human Plks, Plk-1 is overexpressed in many tumor types. Of all mitotic kinases, Plk1 is often overexpressed, making it one of the most validated targets among mitotic kinases. Experimental results demonstrated that modulating Plk1 activity in both transformed and normal cells has an anti-proliferative effect. Plks are significantly involved in the assembly and dynamics of the mitotic spindle apparatus and in the activation and inactivation of CDK/cyclin complexes. In mammalian cells, Plk1 protein levels increase as cells approach the M phase, with peak phosphorylation activity occurring during mitosis. Known substrates of Plk1 include Cdc25C phosphatase, cyclin B, a cohesin subunit of the mitotic spindle, subunits of the anaphase-promoting complex, and mammalian kinesin-like protein 1 (MKLP-1), among other kinesin-related motor proteins. These substrates underscore Plk1's multifaceted role in promoting mitosis. Plk1 also controls the tyrosine dephosphorylation of CDKs by phosphorylating and turning on Cdc25C.
Cancer cells treated with Plk inhibitors either undergo apoptosis or become committed to mitotic catastrophe. Conversely, non-transformed proliferating cells are reversibly arrested at the G2/M boundary. Small-molecule Plk inhibitors have demonstrated selective anti-proliferative effects on cancer cells, producing phenotypes consistent with the established functions of Plks.
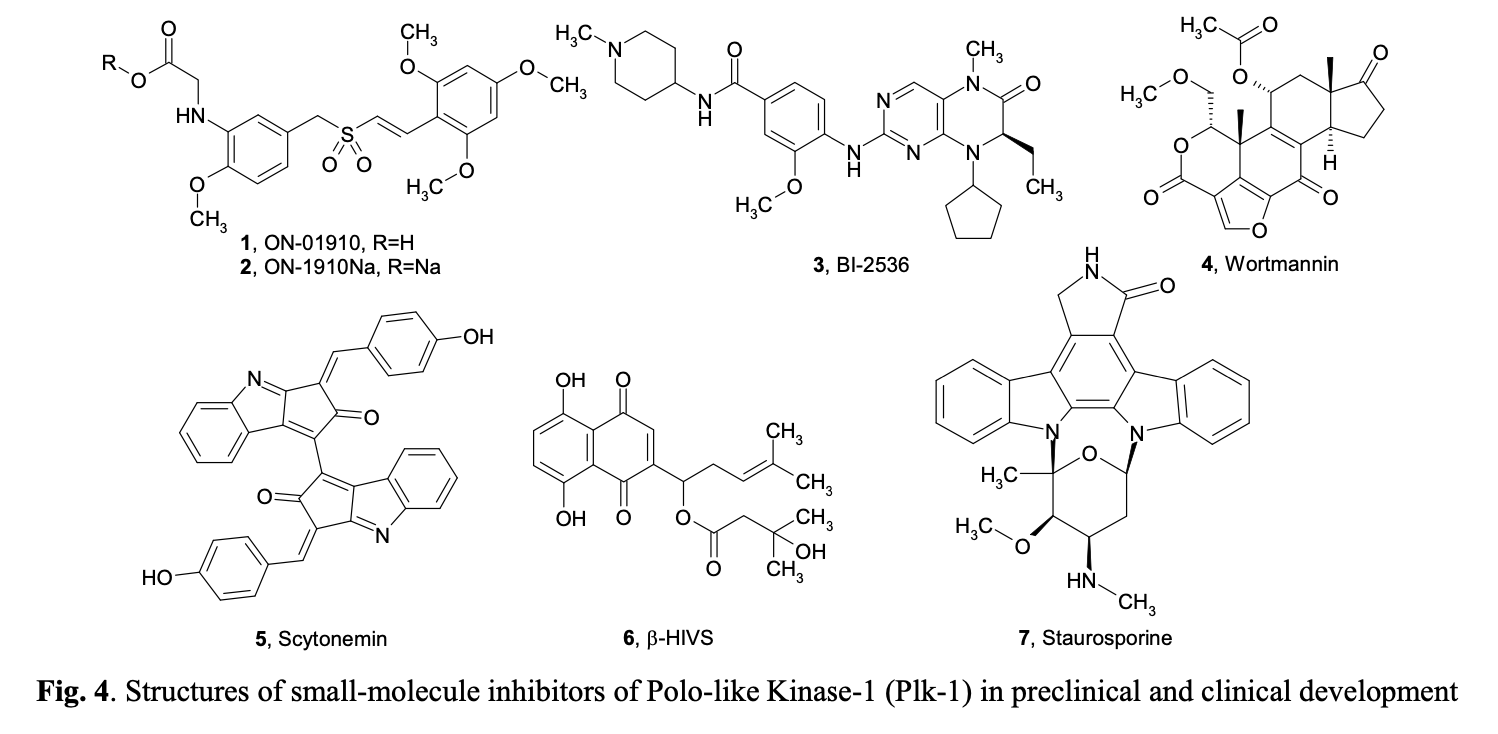
Aurora kinase inhibitors
Serine/threonine protein kinases from the Aurora family play a crucial role in chromosome segregation and cell division across all eukaryotes. Initially identified in the cell cycle studies as Xenopus Eg2, those enzymes are now known as vital components of the spindle checkpoint system used by cells to ensure the fidelity of mitosis. Deregulation of Aurora kinases can disrupt spindle assembly, checkpoint function, and cell division, leading to chromosome missegregation or polyploidization accompanied by centrosome amplification. All Aurora kinases share a similar structure: their catalytic domains are flanked by very short C-terminal tails and N-terminal domains of variable lengths. Given that Aurora kinases regulate the progression of the mitotic cycle at multiple stages (refer to Fig. 2), they are believed to influence numerous proteins. For instance, Aurora A phosphorylates several targets, including Histone H3 (S10), the KSP motor protein, CPEB, PP1, D-TACC, and TPX2. Aurora B regulates the activity of proteins like Histone H3 (S10/S28), CENP-A, INCENP, REC-8, MgcRacGAP, Vimentin, GFAP, and Desmin. Aurora A localizes in centrosomes from the S/early G2 phases and is essential for establishing a bipolar mitotic spindle. Aurora B is associated with chromosomes in early mitosis and then migrates from centromeres to microtubules at the spindle equator in late mitosis. As the spindle elongates and the cell undergoes cytokinesis, Aurora B accumulates in the spindle midzone prior to its final concentration at the midbody. All members of the Aurora kinase family are exclusively expressed during mitosis.
At least two isoforms, specifically Aurora A and B, are commonly overexpressed in human tumors, as seen in primary colon tumor samples. Further studies have suggested that they play a pivotal role in the development of breast, colorectal, bladder, and ovarian cancers (Table 3).
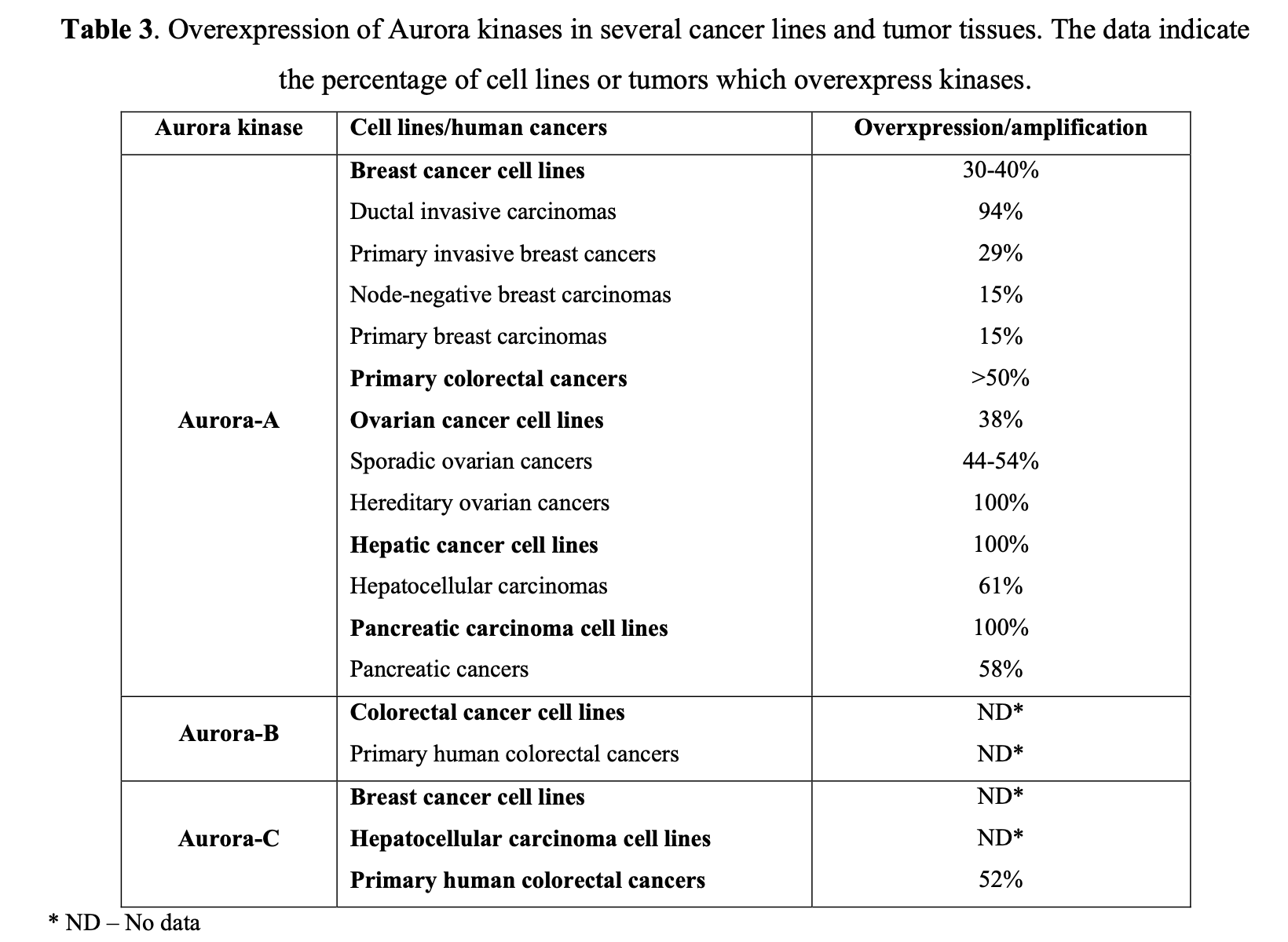
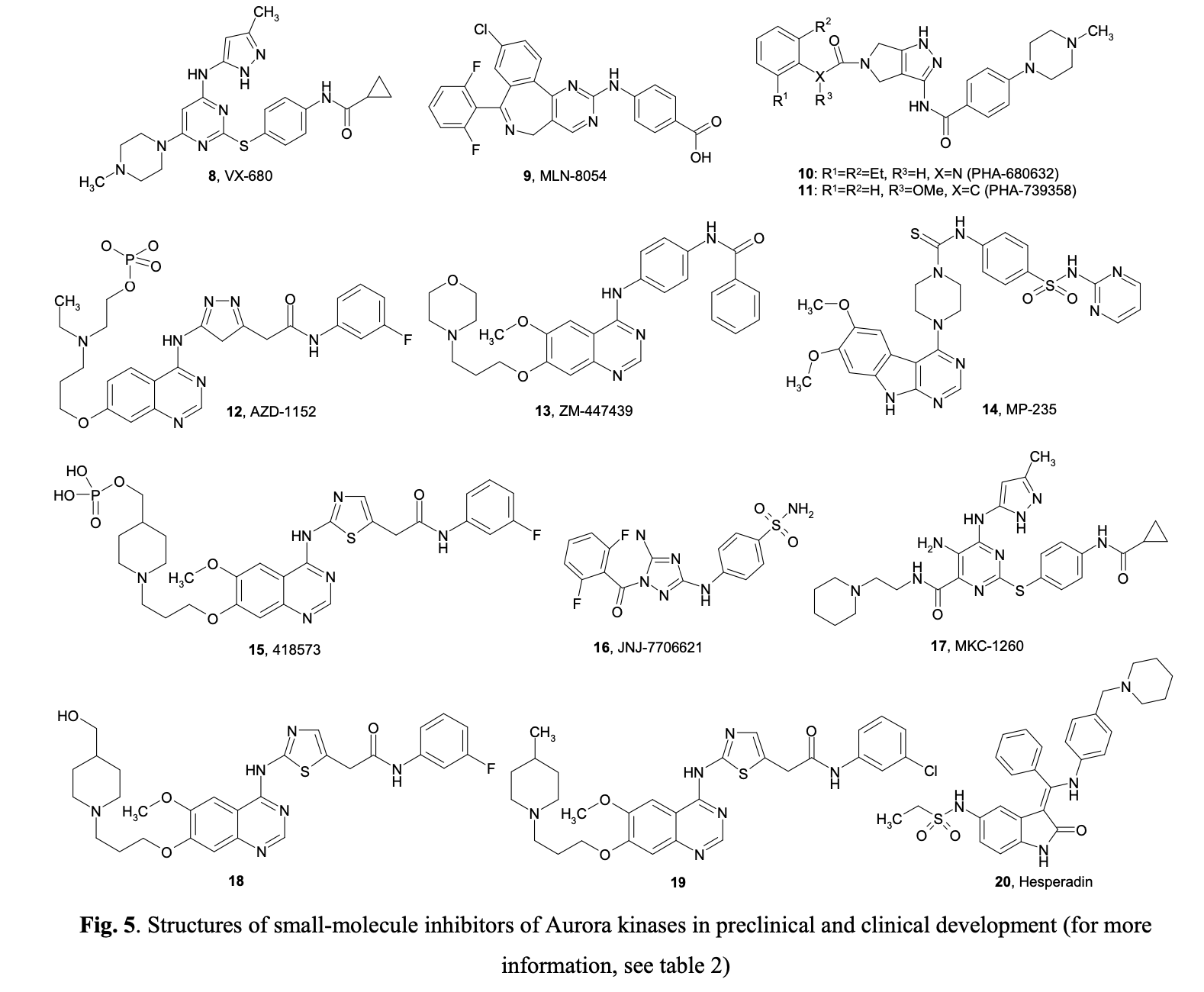
Aberrant expression of Aurora-A kinase leads to genetic instability, either through abnormal centrosome duplication or defects in the spindle checkpoint. Similarly, misregulated levels of Aurora-B can cause abnormalities in chromosome attachment or alignment to the mitotic spindle during cellular mitosis. Aurora-B may form complexes with Survivin, an anti-apoptotic protein commonly overexpressed in tumors. It has been suggested that overexpression of Aurora B may help protect tumor cells from apoptosis. Following the discovery that Aurora kinases are upregulated in many tumors, several small molecule inhibitors with a high level of selectivity to Aurora kinase proteins have been developed. Figure 5 summarizes the structures of selected compounds.
Inhibitors of Cyclin-dependent kinases
Inhibitors of Cyclin-dependent kinases (CDK)
Cyclin-dependent kinases (CDKs) are part of a large family of serine/threonine kinases that play a crucial role in cell cycle regulation, in particular at the early stages of mitosis. They are also involved in the regulation of transcription and mRNA processing. However, CDK9 is the only exception, as it does not participate in cell cycle regulation. As serine/threonine kinases, CDKs phosphorylate proteins on serine and threonine amino acid residues. A cyclin-dependent kinase is activated by forming a complex with a cyclin. The subfamily of CDKs includes several classes, named CDK1–9. A cyclin-CDK complex is typically regulated by various kinases and phosphatases, including Wee, CDK-activating kinase (CAK), and Cdc25. CAK adds an activating phosphate to the complex, while Wee contributes an inhibitory phosphate; the presence of both activating and inhibitory phosphates renders the complex inactive. Cdc25 is a phosphatase that removes the inhibitory phosphate added by Wee, thereby activating the whole complex. CDKs trigger feedback to Wee and Cdc25, inhibiting and enhancing their respective activities.
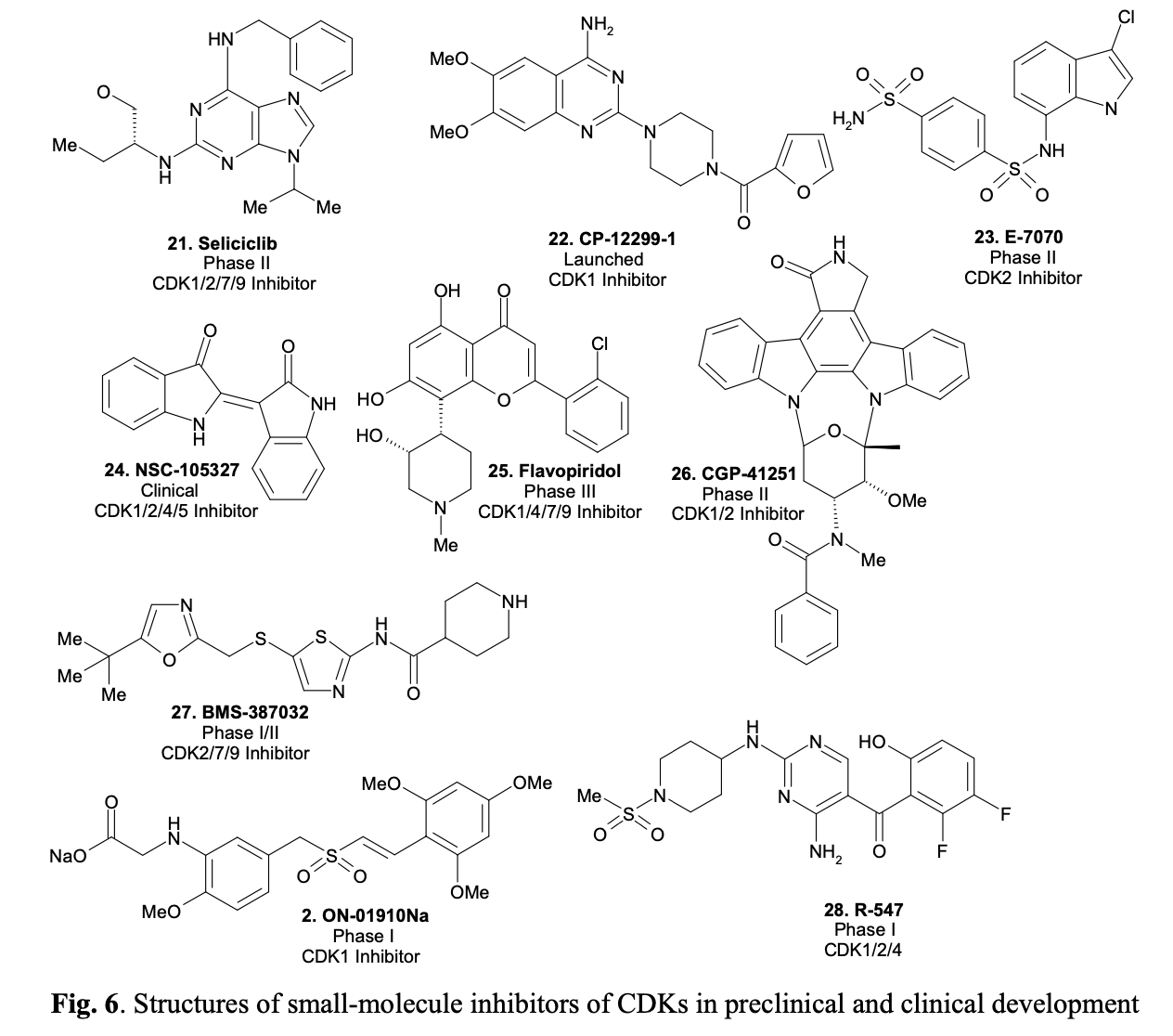
CDKs are known today as potential targets for anti-cancer therapeutics. By selectively disrupting cell cycle regulation in cancer cells through interference with CDK action, those cells are targeted for programmed death. Currently, several CDK inhibitors, such as Seliciclib, are undergoing clinical trials. Originally developed as a potential anti-cancer drug, Seliciclib (shown in Fig. 6) has also been found to induce apoptosis in neutrophil granulocytes, which mediate inflammation, as shown in recent laboratory tests. This discovery opens up the possibility of developing novel drugs for the treatment of chronic inflammatory diseases such as arthritis or cystic fibrosis. Representative structures of small-molecule inhibitors of CDKs, either in preclinical and clinical trials or even on the market, are given in Figure 6.